
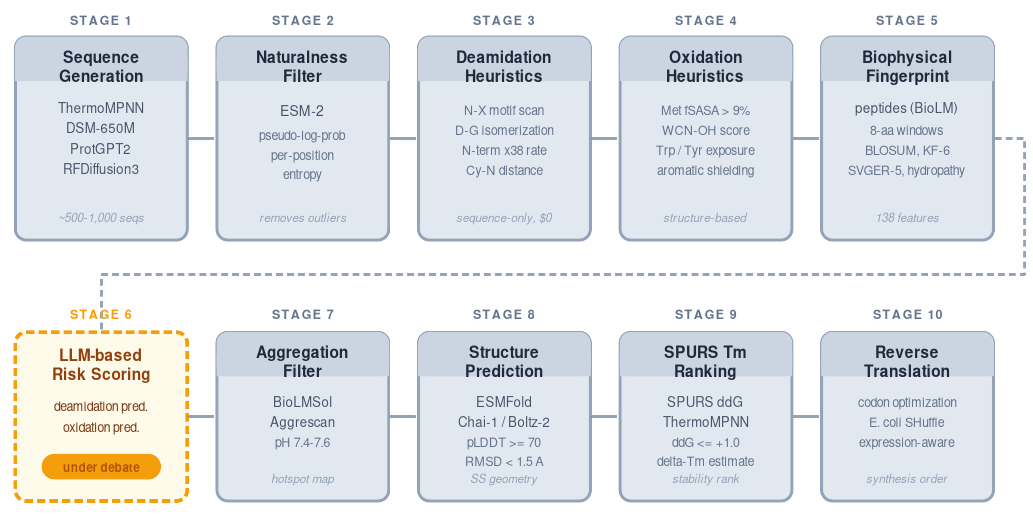
Protein models are more available than ever. That has not made molecular design easy. What matters is knowing what problem you are actually solving, which model outputs are trustworthy on that target, and how to turn all of that into a design process that can survive contact with real biology.
Two years ago, we learned something uncomfortable: giving people access to more protein models did not make molecular design easier.
We kept seeing the same failure modes. Teams optimized melting temperature when the real problem was formulation instability. They generated too few candidates, or too many that were all biased in the same direction. They treated pseudo-log-probabilities and confidence scores as answers instead of signals. They assumed that if a model worked well in one setting, it would work well enough on their target.
That is rarely how these projects fail. The first mistake in protein design is often upstream of generation.
The industry still overestimates how much value comes from access to foundation models and underestimates how much comes from target-specific benchmarking, model calibration, and experimental context. Access to models was never the hard part. The hard part is knowing what problem you are actually solving, which model outputs are trustworthy on that target, and how to turn that into a design process that can survive contact with real biology.
At BioLM, that has pushed us toward a very specific way of working. We do not start with “Which model should we run?” We start with “What is actually failing, what would success look like in the assay, and what evidence would justify trusting a ranking before we spend synthesis slots on it?”
In one recent small, disulfide-rich formulation-stability program, that distinction mattered. The project could easily have been framed as a generic thermostability exercise. It was not. Once we dug into the target, formulation context, and assay requirements, it became clear that the design problem was broader: chemical liabilities, colloidal behavior, structural constraints, and preservation of function all had to be handled together. The pipeline only became clear after the discovery work.
What follows is the process we use before we trust a generative design workflow.
Where molecular design still goes wrong
There are a few recurring ways these projects go off track.
The first is optimizing the wrong objective. A team says they want a “more stable” molecule, but stability can mean several different things: intrinsic fold stability, oxidation resistance, reduced deamidation, lower aggregation risk, improved expression, or simply better behavior in the formulation or assay that matters commercially. If you optimize the wrong proxy, you can get a cleaner score and a worse molecule.
The second is assuming model fit instead of testing it. We do not assume a model that works in general will work on this target, in this regime, for this objective. Different models behave differently across fold classes, protein lengths, mutation types, and readouts. We have seen sequence log-probability models correlate cleanly with some degradation modes, while dedicated thermostability predictors contribute very little signal on out-of-distribution folds. We have also seen structure predictors look confident while recovering the wrong geometry. A score without calibration is just a number.
The third is treating generation as a one-model sampling problem. Once a target is benchmarked, generation is not “pick a favorite model and sample.” Different generators explore different parts of sequence space. Some are conservative and stay close to the wild type. Some aggressively trade naturalness for a particular objective. Some are useful precisely because they reveal a competing optimum that other methods will not find. If you only generate one kind of candidate, you learn one kind of lesson.
The fourth is collapsing selection into a single leaderboard. Round 1 is not only about picking the top sequences most likely to win on paper. It is also about building the best possible feedback loop for Round 2. That means maximizing useful variance, controlling mutation burden, keeping some candidates close to natural sequence space, and deliberately preserving disagreement between computational criteria. If every shortlisted sequence looks the same computationally, you may get a clean result set and learn almost nothing.
That is why our process starts before generation.
Sprint 1: Define the real problem, not the proxy
Before we design a single variant, we try to define what is actually failing.
That sounds obvious, but it is where many projects drift. “Increase thermostability” is a tempting brief because it is legible and measurable. But in practice, the molecule may already be thermally stable enough in isolation. The real problem may be oxidation in the formulation, deamidation at specific sites, surface-patch-mediated aggregation, poor expression, or loss of function under a process constraint. If that is true, optimizing generic Tm can make the workflow feel rigorous while solving the wrong problem.
So the first sprint is about problem framing. We gather the target biology, what is known about degradation pathways, what is already fixed by structure or function, what the relevant assay actually measures, and whether current tools and available data make the project feasible in the first place. We do not take on design work just because a target is interesting. We take it on when there is a plausible path from current tools and available data to a useful decision.
On a recent small, disulfide-rich program, this changed the project immediately. The initial problem statement could have been reduced to “make the molecule more stable.” Instead, the discovery phase separated intrinsic fold stability from formulation-driven liabilities and forced us to rank the real risks. That reframing affected everything downstream: what we benchmarked, which models mattered, and what the first experimental round needed to teach us.
Sprint 2: Gather the constraints that actually matter
A design problem is defined as much by the assay, production context, and commercial constraints as by the sequence itself.
We want to know what molecular form is actually being produced, what assay defines success, what expression system matters, how far from a natural or previously accepted scaffold we can move, and which tradeoffs are acceptable. Sometimes the mutation count is not just a technical variable. It is tied to developability, manufacturing, regulatory posture, or the practical reality that if you move too far from a natural protein, you may win a model objective and lose the path forward.
This is also where model discovery becomes more grounded. We do not just ask which models are available. We ask which model outputs are likely to correlate with the objectives that matter in this program. If oxidation is a major risk, we want models and heuristics that say something meaningful about oxidation-sensitive contexts. If structure preservation matters, we want to know which structure predictors remain trustworthy for this fold and length. If expression or naturalness is part of the constraint set, we want to know which evolutionary signals are useful and which are misleading.
That early framing affects generation later. Model temperature, for example, is not just a sampling hyperparameter. On some generators, temperature materially changes edit distance, mutation count, and how far a sequence drifts from natural space. If you do not benchmark that early, you can accidentally generate a library full of candidates that are too mutated to be useful, too conservative to be informative, or too biased toward one objective to produce a good feedback loop.
This is why our discovery phase is not only biological. It is computational and operational as well.
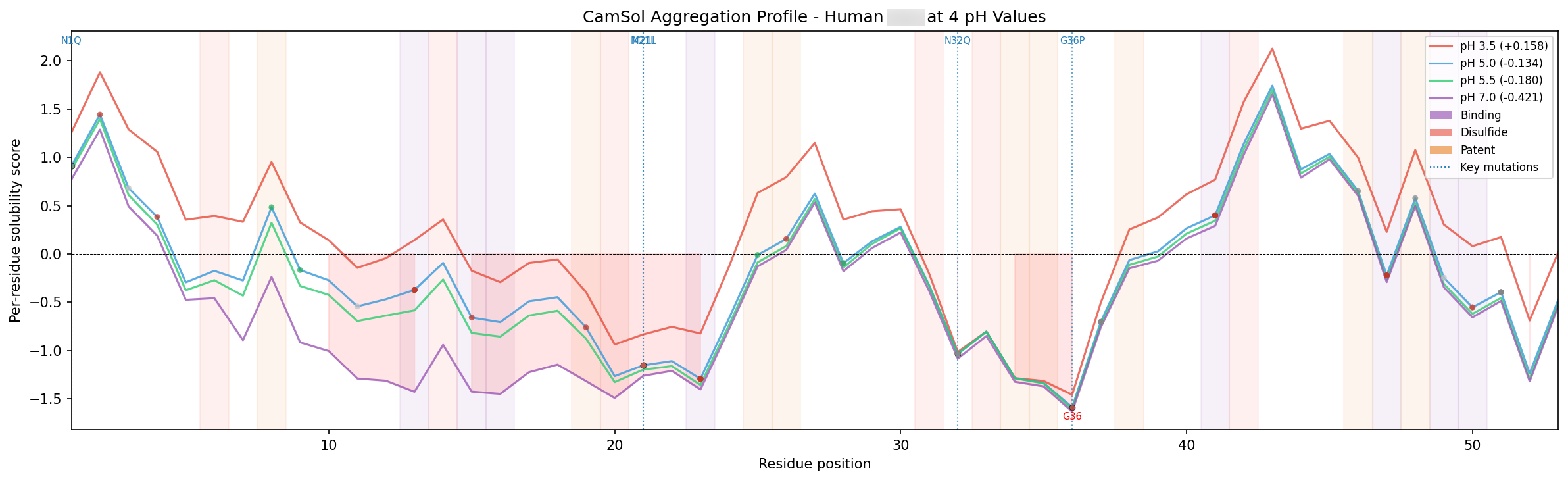
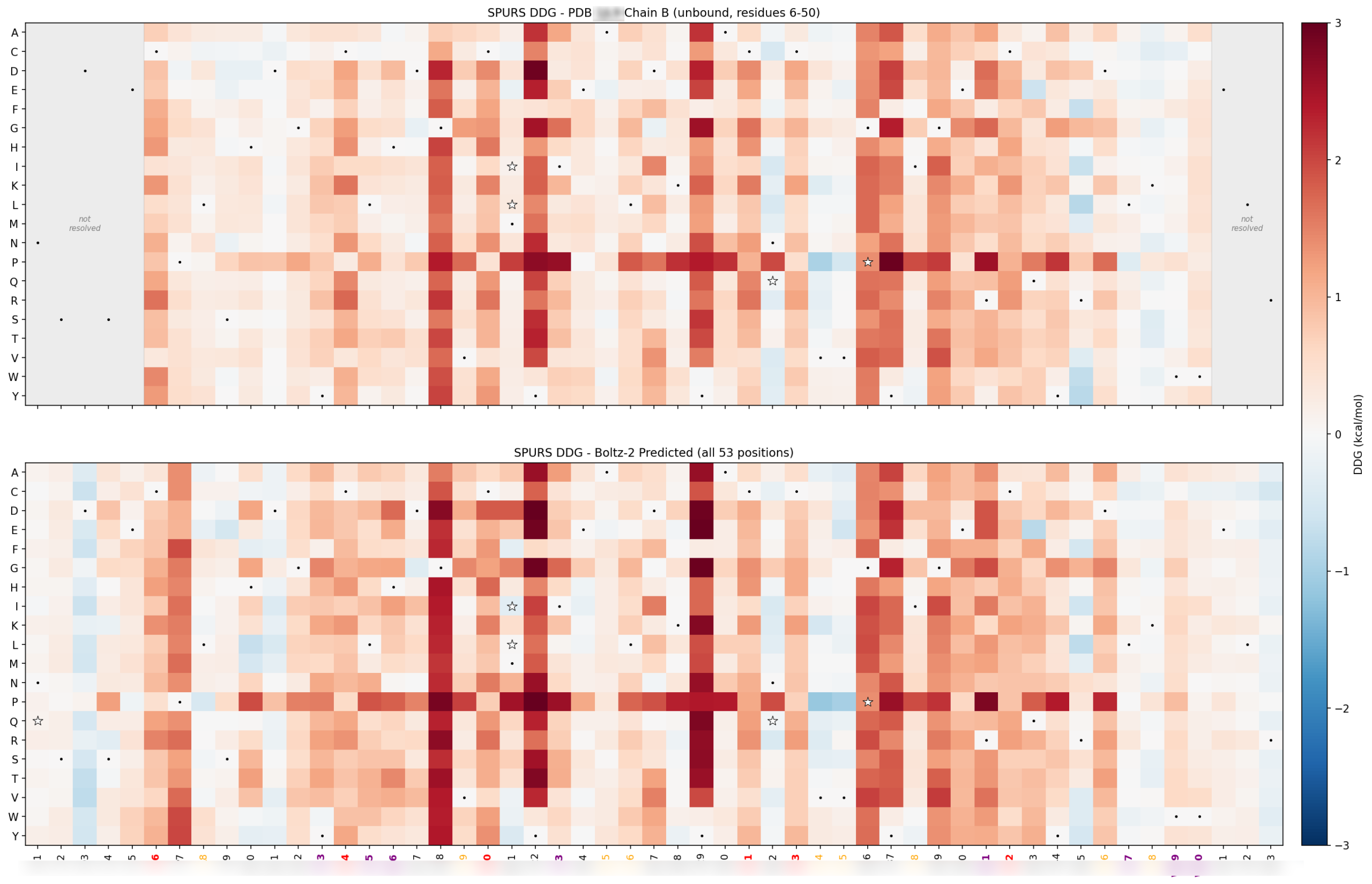
Sprint 3: Build the wild-type baseline before designing anything
Before we generate variants, we want a full computational portrait of the wild type.
That means mapping the target from several directions at once: single-mutation stability landscape, conservation, structural confidence, solubility or aggregation signals, known liabilities, and — when available — known mutations from literature and internally-created datasets. The goal is to build a per-residue map of what the target is likely to tolerate before we ask a generator to propose anything new.
This matters for two reasons.
First, it tells us where the designable space is actually located. Some positions that look mutable on paper turn out to be structurally critical. Others are technically designable but not worth touching because they are too close to a fixed functional constraint. On one recent program, this baseline work made it clear that some intuitively attractive mutations should be dropped very early, while other positions offered dual benefit across more than one liability axis.
Second, the wild-type baseline gives us a reference point for every downstream score. We do not want to rank candidates against each other without first knowing how the underlying models behave on the original scaffold.
This stage is unglamorous, but it prevents a lot of wasted motion. If you skip the baseline, the generator becomes your first exploratory tool. That is backwards. Generation should happen after you understand the surface you are exploring.
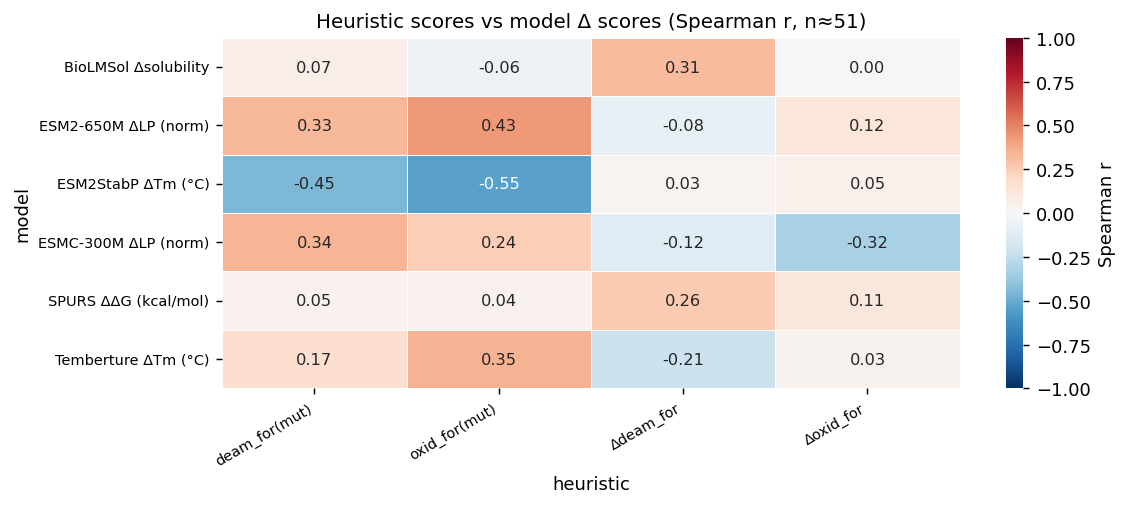
Sprint 4: Benchmark the models before trusting them
This is the step that tends to get skipped, and it is probably the most important.
We benchmark the models on the target before we let them influence ranking. That means retrospective scoring on known variants where possible, structure benchmarking on the actual scaffold, and asking a simple question: which outputs are usable signals for this objective, and which are not?
We have learned to be skeptical here. A model that performs well in one domain may be weak or noisy on a short, disulfide-rich protein. A structure predictor may report high confidence and still recover the wrong fold geometry. A sequence log-probability model may capture one degradation mode surprisingly well while a dedicated predictor contributes no meaningful signal for that task. None of this is theoretical. We see it in practice.
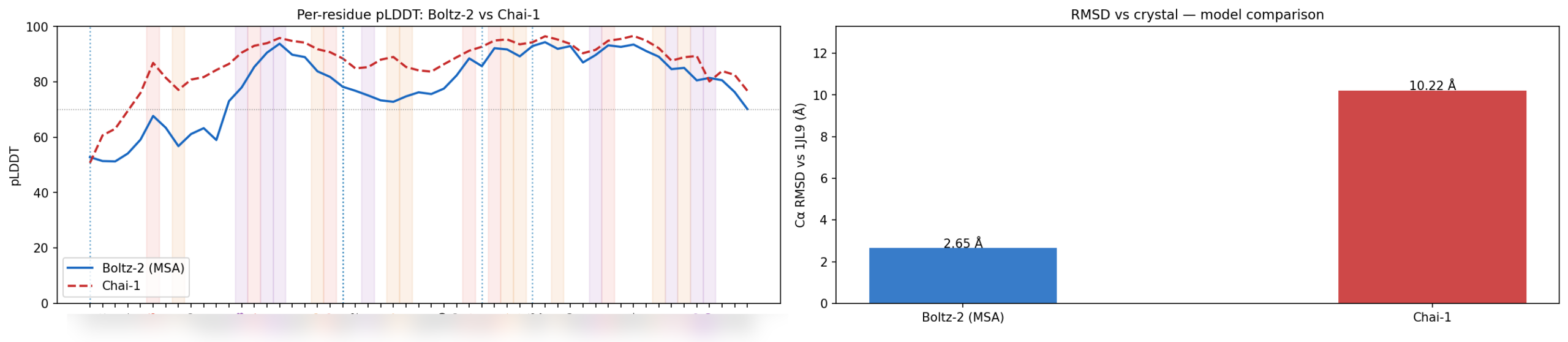
In one recent workflow, structure benchmarking showed that one predictor became meaningfully more trustworthy once it had MSA support, while another could look confident without recovering the correct structure. In broader labeled degradation analyses, some LP-based models showed strong signal for oxidation and deamidation-related outcomes, while some thermal-stability models provided little useful correlation on out-of-distribution examples. That does not mean those models are “bad.” It means they are not equally useful for every target and every decision.
This is why we do not collapse all scores into one composite number at the start. We first decide what each score is allowed to mean.
Benchmarking also changes how we describe the workflow internally. A model can be a generator, a scorer, a filter, a sanity check, or something we deliberately exclude from ranking until it earns trust. Those are different roles. Mixing them together is how pipelines become opaque.
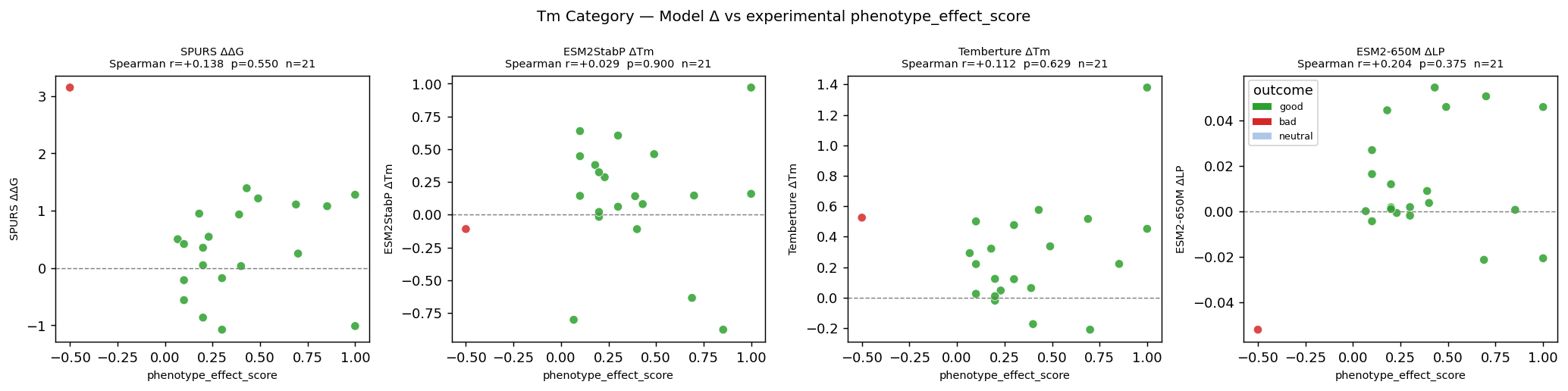
Sprint 5: Treat generation as portfolio design, not one-model sampling
Once the target is benchmarked, generation is not “pick a favorite model and sample.”
It becomes a portfolio design problem.
We want multiple generators because different models explore different parts of sequence space. In one recent scan, one MPNN-family operating point dominated the composite ranking under low-temperature, full-region generation, while a diffusion-based model discovered a very different optimum: higher thermal stability at the cost of solubility. That tradeoff axis would have been invisible in an MPNN-only screen. At the same time, sequential low-Hamming evolutionary sampling produced a different kind of candidate entirely: much closer to natural sequence space, lower variance, and useful for understanding what the model considers genuinely plausible.
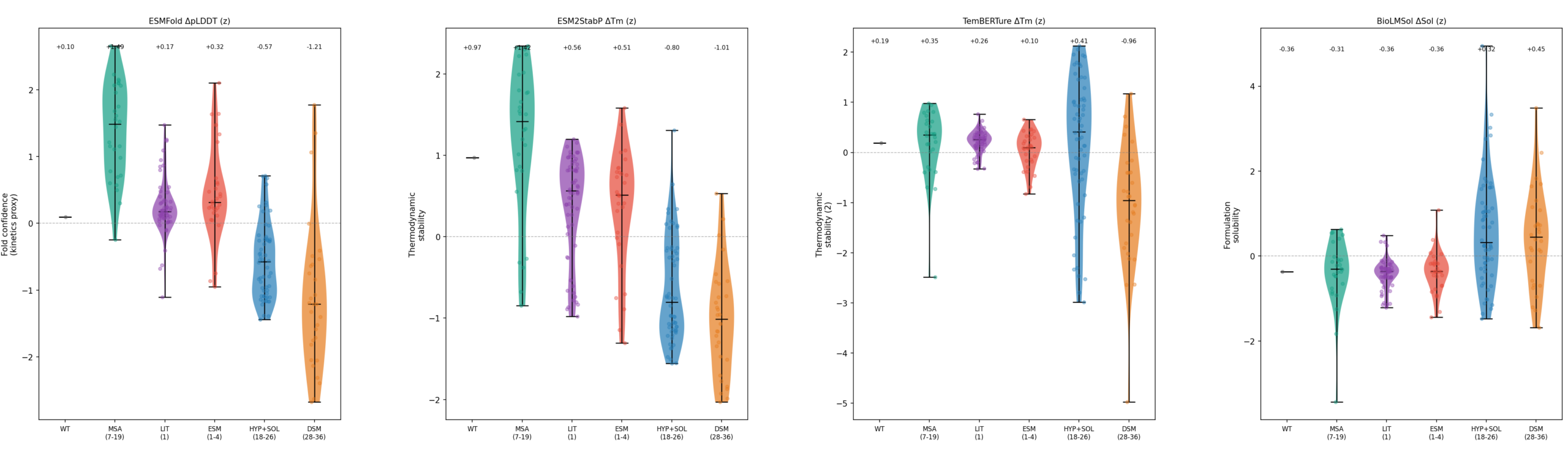
That is exactly why we do not want a single generator deciding the shape of Round 1.
This is also where mutation-count control becomes practical rather than philosophical. We benchmark temperature, aggressiveness, remasking strategy, step size, and design region because those choices directly affect edit distance, mutation burden, and diversity. Temperature scanning is not a decorative sweep. It is how you learn whether a model is going to collapse to near-wild-type sequences, generate the wrong number of mutations, or push too far away from a realistic design space.
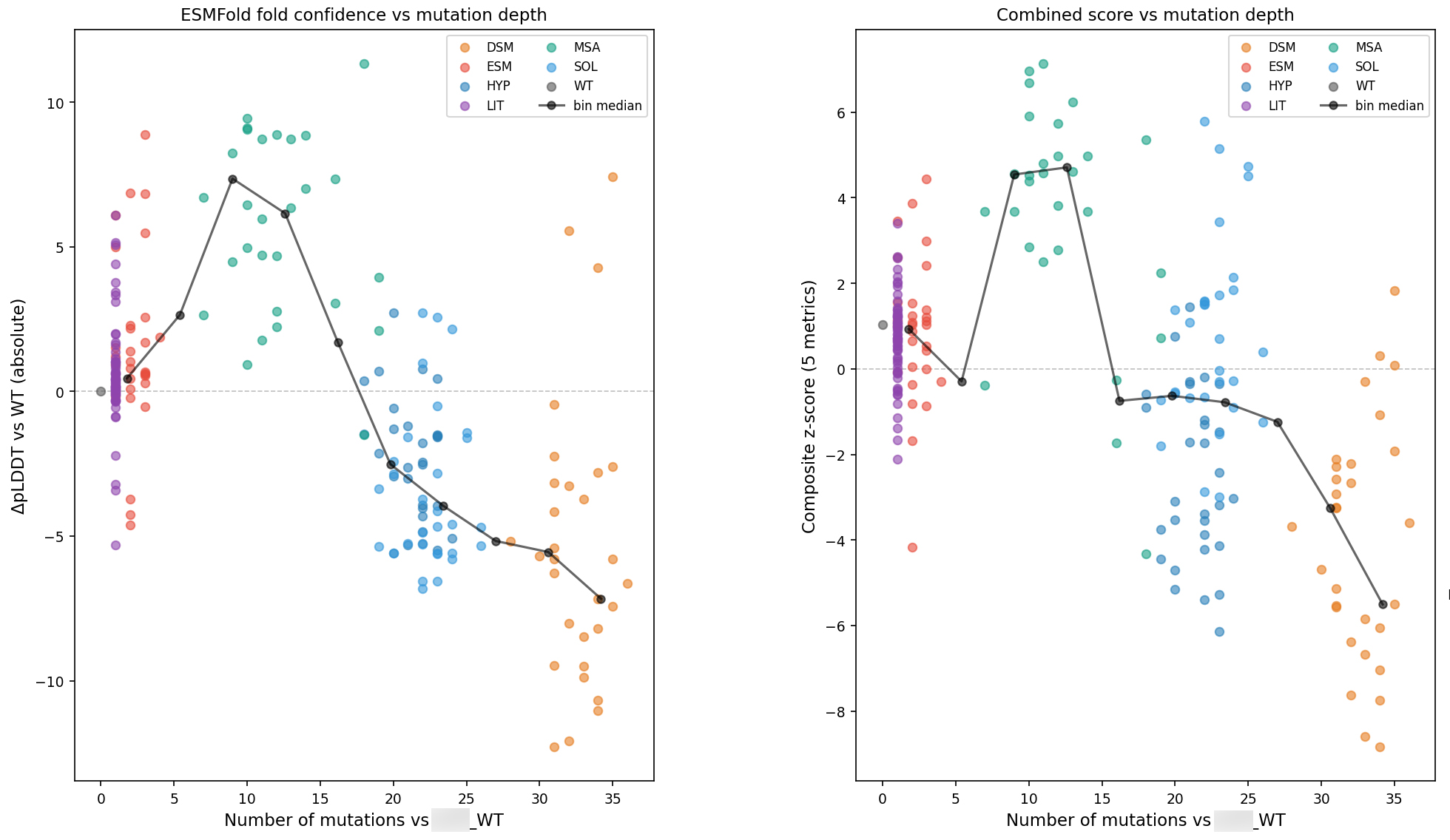
And the “best” setting is not universal across model families. On one recent program, lowering temperature improved one generator’s operating point, while another reached a local trap at the same low setting. Some regional patches underperformed and were dropped. Other settings produced better diversity without improving composite score, which made them useful as deliberate hedges rather than primary cohorts.
That is the kind of selection logic we care about. We are not only maximizing immediate score. We are designing a library that can teach us something in the next round.
So when we shortlist sequences, we do not simply take the top dozen by one metric. We preserve candidates that are strong under different, sometimes competing criteria: top composite candidates, stability-focused candidates, low-Hamming natural candidates, diversity hedges, and candidates that are “safe” under heuristic degradation screens. That keeps the feedback loop alive. If every chosen sequence wins the same way computationally, the wet-lab result will tell you less than you think.
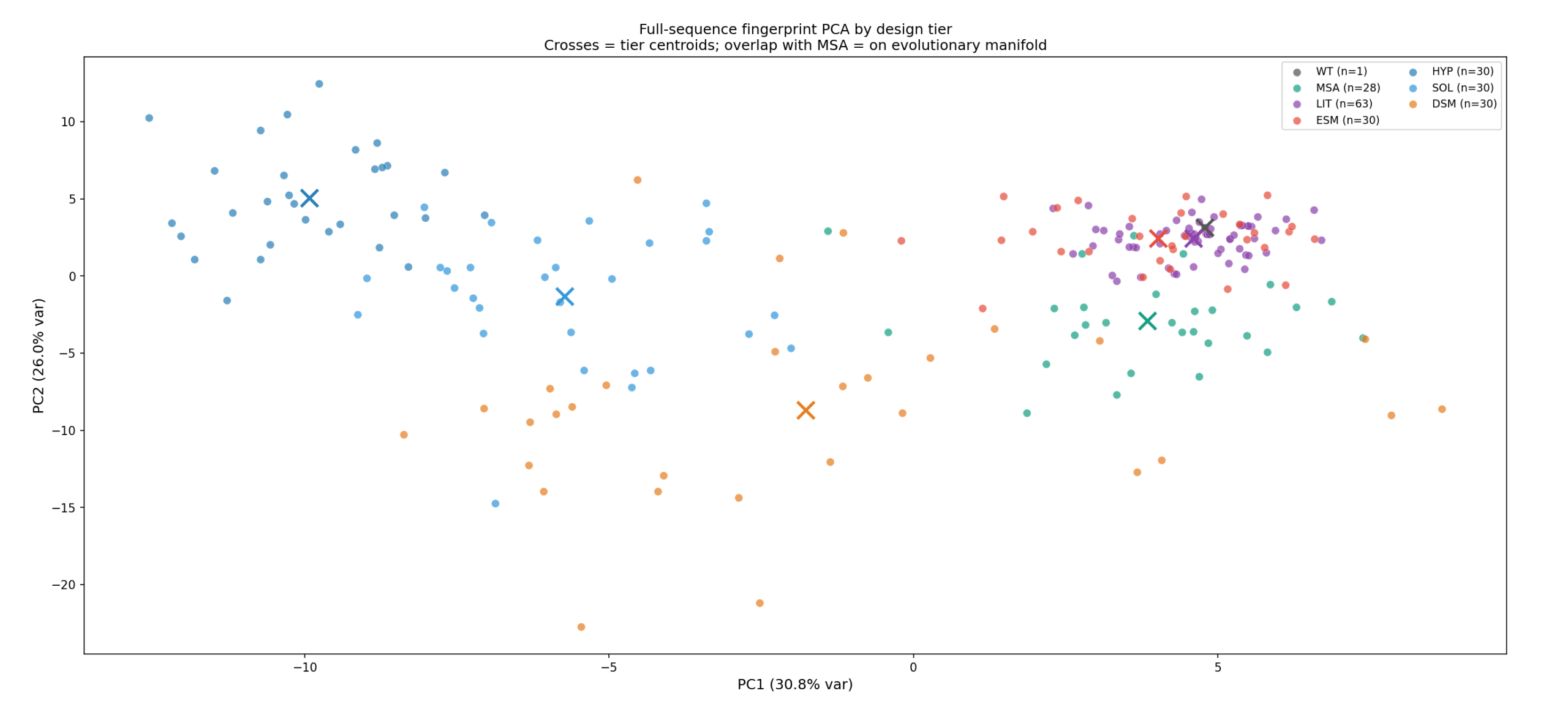
Sprint 6: Use cheap filters before expensive ones
Not every decision deserves a GPU.
Once we have the portfolio of generated candidates, we try to remove obvious liabilities cheaply before we spend expensive model calls on them. That means motif-based filters, chemistry-aware heuristics, sequence-only fingerprinting, and fast structural or naturalness proxies where they make sense. Only after that do we run the more expensive structure, complex, and multi-objective scoring stages.
This is partly about efficiency, but mostly about discipline. If a candidate fails an obvious degradation heuristic or pushes a sequence-level fingerprint far into a region associated with structural collapse, we do not need to pretend that the expensive model call made the workflow more rigorous. It just made it slower.
The interesting part is that these cheap filters are not only subtractive. They are also informative. In our recent work, sequence- and window-level fingerprints carried meaningful signal about downstream stability behavior. They also helped differentiate generator families and quantify when a candidate pool was drifting into an unproductive region of sequence space. That makes them useful both as prefilters and as tools for designing future rounds.
This is where builder discipline and scientific judgment meet. The cheapest useful filter should come first. The expensive models should spend time on candidates that still matter.
Only after all of that do we draw the pipeline.
By then, the workflow is no longer a generic sequence of model calls. It is a target-specific process with reasons behind every stage: what was benchmarked, what was filtered cheaply, what was calibrated, what was retained for diversity, and what was excluded from ranking until proven useful.
That is the point of the pipeline. It is not the starting point. It is the output of the discovery process.
The pipeline is the result, not the starting point
By the time we draw the workflow, most of the important thinking has already happened.
The final pipeline is not just a collection of model calls. It is the operational summary of the discovery phase: what problem we are actually solving, which models earned trust on the target, how much mutation burden is acceptable, where diversity needs to be preserved, and which cheap filters can reduce noise before expensive scoring starts.
That is what makes a pipeline useful. It is not just reproducible. It is justified.
What this changed for us
The biggest lesson for us over the last two years has been simple: access to models is not the same as access to successful molecular design. We are most interested in how we can enable successful use of molecular modeling, not simply provide our users access to the tools.
Remember that what matters is the framing, the calibration, the constraint mapping, the mutation-budget logic, the portfolio design, and the discipline to decide what a score actually means before you act on it. It is also the willingness to design Round 1 for Round 2: to preserve variance, keep useful disagreement between computational criteria, and build a better learning loop rather than a cleaner slide.
That is not as marketable as saying “we have access to the latest models.” But this is the part that decides whether the computational workflow is useful.
If your team has a target in mind, the first question usually is not “which model should we run?” It is “what problem are we actually solving, and what would convince us we are solving it?” That is where we start. If you are looking for advice, reach out solutions@biolm.ai with any questions about your own projects.