
The Notebook as Foundation: Drafting Your First Protocol
Notebooks are a very useful way to draft a BioLM protocol the first few times you run through it. They allow for interactive exploration, quick iteration, and the flexibility to experiment as you work through a problem. That’s exactly how our antibody engineering workflow started: as a Jupyter notebook demonstrating how to process solved antibody–antigen complexes, extract heavy and light chains, and apply BioLM’s AntiFold model to redesign the CDRs.
The notebook walked through four classic targets from the BenchBB dataset—IL7Rα, PDL-1, EGFR, and MBP—showing how BioLM can generate and evaluate variant binders. As an exploratory document, it did its job well: guiding readers through parsing PDB structures, setting design regions, and generating candidate variants. The notebook successfully demonstrated a complete pipeline from raw protein structures to final analysis results.
However, as useful as notebooks are for drafting protocols, eventually with enough repetition you want to formally define your protocol into a workflow. When you need to run the same analysis across multiple targets, share it with colleagues, or extend it to new use cases, the notebook’s limitations become apparent. The linear structure makes it difficult to modify individual components, hardcoded parameters make experimentation cumbersome, and the lack of clear data dependencies makes debugging challenging.
Kedro is an ideal framework for making exactly this transition. It provides a way to move from linear, notebook-based code into modular, reproducible pipelines. With its data catalogs, configuration management, and pipeline visualization tools, Kedro turns exploratory work into a system that is easier to maintain, reuse, and share.
The Translation Process: From Linear Code to Modular Pipelines
Here we present an example of how to transition from notebook to Kedro pipeline by systematically restructuring a workflow to take advantage of the framework’s capabilities. The first step involves understanding the notebook’s structure and data flow. We mapped out each major operation: protein structure parsing, calls to BioLM models, variant analysis, and visualization generation. These components serve as the obvious groupings of Nodes in Kedro that we can translate our notebook code into.
With a clear understanding of the workflow, we designed a pipeline architecture that separated concerns into distinct phases: raw data processing, primary analysis, and reporting. Each phase had well-defined inputs and outputs, making the data flow explicit and easier to understand. We broke down the notebook’s functionality into discrete, focused functions—each with a single responsibility like extracting protein chains, generating variants, analyzing results, or creating visualizations. This modular approach made the code more testable and maintainable.
Much of what looked like “code” in the notebook was actually configuration—file paths, model parameters, and protein target definitions. Kedro’s configuration system allowed us to abstract these settings into its YAML based configuration files, making the system more flexible and easier to modify on subsequent iterations. We defined all data inputs and outputs in Kedro’s data catalog, which provides a centralized registry of datasets with their types, locations, and metadata. This approach makes data dependencies explicit and enables features like data versioning and lineage tracking.
The final step was assembling the functions into a Kedro pipeline using the framework’s node-based architecture. Each function became a node with clear inputs and outputs, creating a directed graph that represents the complete data flow.
Running the Pipeline and Customizing Experiments
Once the Kedro pipeline is set up, running the complete antibody engineering workflow is straightforward. The pipeline can be executed with a single command: kedro run --pipeline antibody. This processes all four protein targets (EGFR, PDL1, MBP, and IL-7Rα) through the complete workflow, from raw PDB structures to final variant analysis.
Results are organized in the data/ directory structure, with intermediate outputs in 02_intermediate/, processed variants in 03_primary/, feature engineering results in 04_feature/, and final reports and visualizations in 08_reporting/. The pipeline produces the same outputs as the original notebook but in a more structured format: individual variant CSV files for each protein target (EGFR, PDL1, MBP, IL-7Rα), a comprehensive dataset combining all variants with CDR annotations, and analysis visualizations including CDR sequence distribution plots and variant pairplots that show the relationships between different scoring metrics.
One of the key advantages of the Kedro approach is the ability to easily customize experiments through configuration files. The conf/base/parameters.yml file contains all the configurable parameters, including protein target definitions, API settings, and analysis parameters. To run the pipeline on different protein targets, simply modify the target list in the parameters file without touching any code. Now, you can dynamically change these parameters at run time (from the command line, for example), but the “Kedro way” is really to define and commit the encoded parameters directly into version control.
The pipeline also supports running from different checkpoints, which is particularly useful for long-running analyses or when you want to focus on specific parts of the workflow. For example, you can run only the variant generation step with kedro run --from-nodes generate_variants or start from a specific intermediate result with kedro run --from-outputs processed_variants. This checkpoint functionality makes it easy to iterate on specific parts of the analysis without rerunning the entire pipeline.
Making the Workflow Accessible and Reproducible
Once the pipeline was structured, we focused on making the workflow accessible to others and ensuring reproducibility. We set up GitHub Actions workflows that automatically test and run the pipeline, then generate and deploy a Kedro-Viz visualization to GitHub Pages whenever changes are made.
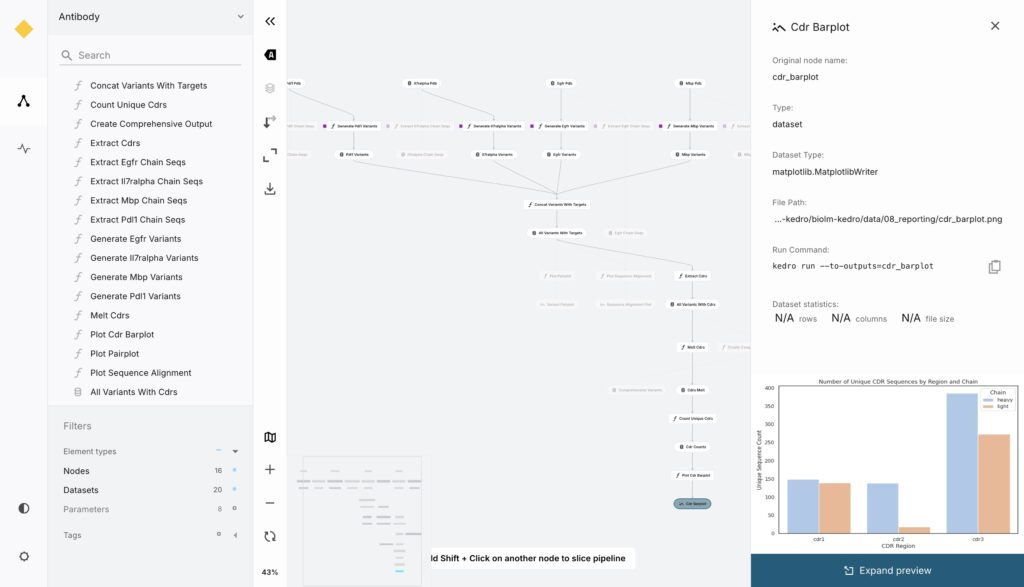
Kedro-Viz is Kedro’s built-in visualization tool that creates an interactive web application showing the complete pipeline structure. It displays the data flow as a directed graph with nodes representing each processing step and edges showing data dependencies. Users can click on any node to see its inputs, outputs, and execution metadata, making it easy to understand how data moves through the workflow.
The deployment process builds this Kedro-Viz application and publishes it to GitHub Pages, creating an automatically updating system that reflects the current state of the pipeline. We also enabled dataset previews, allowing users to see sample data at any point in the pipeline. This required configuring Kedro-Viz with the --include-previews flag and properly setting up metadata in the data catalog. The result is a transparent system where anyone can understand the workflow and inspect intermediate results without needing to run the pipeline themselves.
Key Takeaways and Framework Benefits
This example demonstrates Kedro as a useful framework for defining BioLM protocols and transforming experimental notebooks into structured, reproducible workflows. The workflow illustrates how Kedro can systematically structure BioLM’s AI-driven biology research capabilities for model-assisted molecular design, making these analyses reproducible and shareable.
A key insight is that much of what appears to be “code” in notebooks can often actually be considered configuration—file paths, parameters, and target definitions. Abstracting these into configuration files makes the workflow more flexible and easier to modify without touching the core logic. This is particularly valuable when working with BioLM’s API, as it allows researchers to easily experiment with different molecular targets, design parameters, and AI model configurations.
Kedro’s strengths for bioinformatics workflows include its data catalog system, which makes data dependencies explicit and enables dataset previews, and its built-in visualization capabilities through Kedro-Viz. The framework’s gradual migration path from notebooks means you can start with existing BioLM protocols and refactor incrementally. The checkpoint functionality allows you to run specific parts of the analysis without rerunning everything, which is particularly valuable for long-running bioinformatics pipelines that involve multiple AI model calls.
The principles we applied—modular design, externalized configuration, and clear data flow—are applicable beyond this specific project. Whether you’re working with protein structures, genomic data, or other biological datasets, these approaches can help improve the reproducibility and shareability of your analysis workflows. For researchers leveraging BioLM’s AI-driven biology platform, this structured approach makes it easier to compare results across different experiments, iterate on molecular designs, and share protocols with collaborators.
For more information about this project, visit the GitHub repository or explore the live Kedro-Viz deployment at https://biolm.github.io/biolm-kedro/.